
 PARENTING TIPS
PARENTING TIPS PREGNANCY
PREGNANCY BABY CARE
BABY CARE TODDLERS
TODDLERS TEENS
TEENS HEALTH CARE
HEALTH CARE ACTIVITIES & CRAFTS
ACTIVITIES & CRAFTSZika virus infections have become a local epidemic in the Americas at various time points in the recent past. A recent study attempted to explore whether this was due to increased transmissibility characteristics due to mutation or recombination.
 Study: Genomic and phenotypic analyses suggest moderate fitness differences among Zika virus lineages. Image Credit: NIAID
Study: Genomic and phenotypic analyses suggest moderate fitness differences among Zika virus lineages. Image Credit: NIAID
Introduction
The Zika virus infected approximately 100 million people during the 2014 epidemic in the Americas. It is known to cause microcephaly in 7% of newborns born to mothers infected during pregnancy, with neurological or ocular defects in another six percent.
These effects were reported only from this time onwards. This led to the hypothesis that “the virus acquired mutations that enhanced virulence” and that it “adapted to mosquitoes or humans, which may have facilitated its explosive spread across the Americas.”
The current study, published in the journal PLOS Neglected Tropical Diseases, aimed to explore the possible phenotypic adaptation of the Zika virus to increase its transmissibility and virulence in the Americas.
First, the scientists analyzed the genomic sequences of the Zika viruses from an array of samples taken during the 2015-16 epidemic. Then, they identified lineages that could have emerged during this time, looking for differences in fitness.
The researchers also produced growth curves for Zika virus lineages by using twelve modified Zika viruses with recombinant genetic material to represent specific lineages of the virus. These viruses infected human cells of several lines, as well as live Aedes aegypti mosquitoes, which are the main vector for this virus.
Many of the mosquitoes fed with infectious blood meals containing one of the clades did not show the presence of the virus in their saliva, perhaps because some of the clades had low titers of the virus, limiting the concentration of virus in all the blood meals. To compensate, the researchers used intrathoracic injections of each of the 12 clades of Zika virus into female mosquitoes.
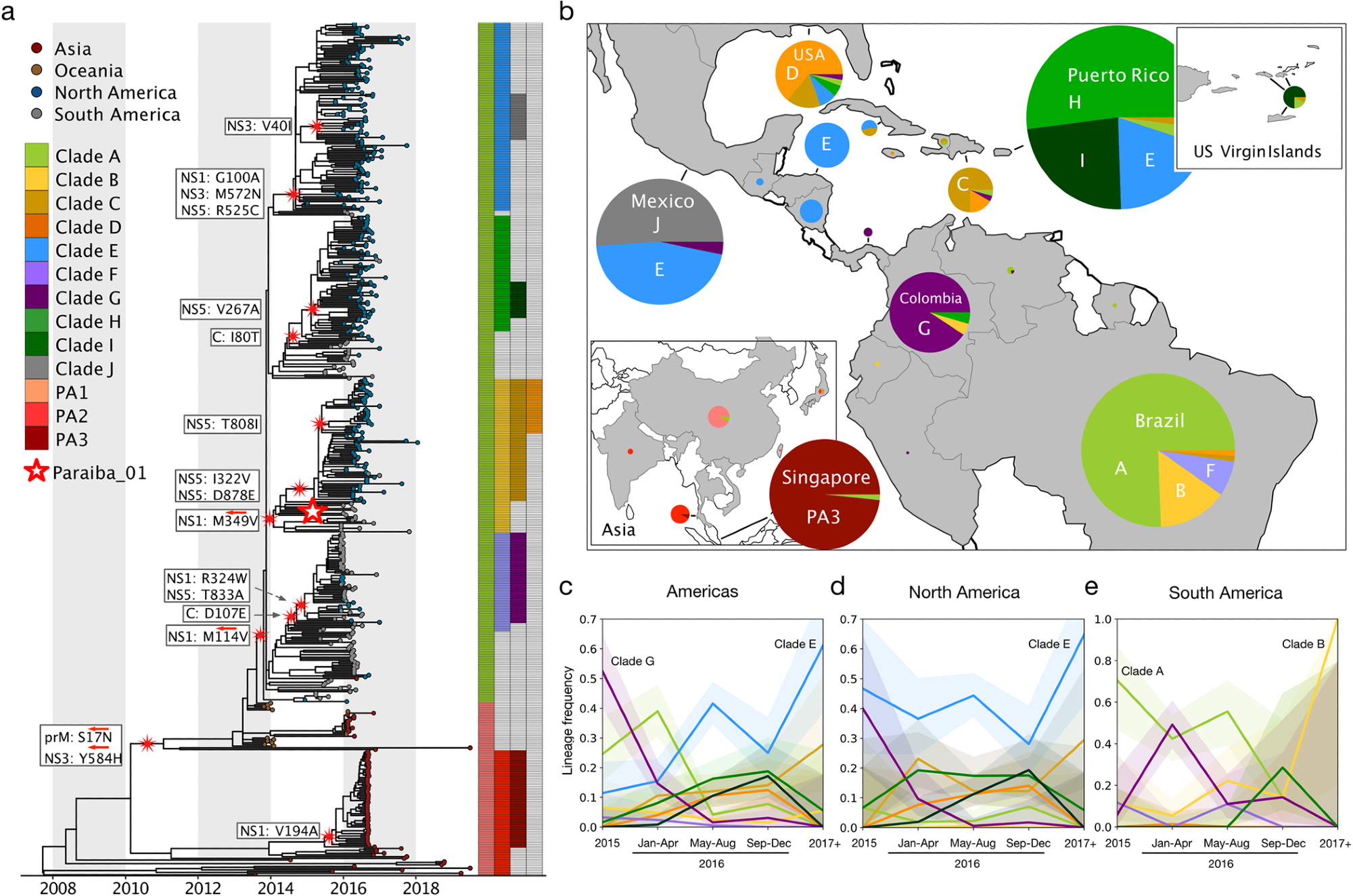
13 major Zika virus lineages defined by nonsynonymous mutations. (a) Zika phylogeny partitioned into 13 lineages defined by nonsynonymous mutations. Large red and white star: location of the initial infectious clone into which specific mutations were introduced. White boxes: nonsynonymous mutations introduced into the initial infectious clone to model each lineage. Red arrows: lineage-defining nonsynonymous mutations that were reverted to their ancestral states. (b) Proportions of clades circulating in countries across the Americas and Asia. Pie sizes represent the number of sequences. (Map made with the coastlines basemap from Natural Earth.) Temporal frequencies of sequenced ZIKV isolates across the Americas (c), North America (including the Caribbean and Central America) (d), South America (e) with 95% confidence intervals. Timepoint 2015- represents isolates collected during or before 2015. Timepoint 2017+ represents isolates collected during or after 2017.
What did the study show?
The study results show that 13 significant clades may have arisen during the 2015-16 Zika virus epidemic, eventually finding thirteen clades based on 17 unique mutations. Of these, clade E seems to have risen in frequency, but none were dominant enough to become fixed.
The infecting viruses gave rise to two lineages, clade B and clade E, with increased replicative fitness following infection in human primary cells. No corresponding divergence was observed within infected mosquitoes. Both methods of mosquito infection failed to show any transmission differences between any of the clades.
Notably, “none of our Zika virus lineages with enhanced replicative fitness displaced ancestral lineages during the epidemic.”
Increased replicative fitness was associated with clade E and clade B mutations, specifically NS1-G100A, NS3-M572N, and NS5-R525C in clade E and (NS1-M349V in clade B. The association between the clade E mutation NS1-G100A and better replicative fitness was earlier reported by other researchers in mouse cells, but its corresponding role in human cells should be validated in future studies.
Clade E is found in most parts of Central America, while clade B was an early Brazilian variant.
The prM-S17N substitution differentiates the PA2 lineage from clade A Zika virus but does not explicitly confer fitness or virulence on the virus. However, since the NS3-Y584H mutation also defines the PA2 lineage, the independent effects of these mutations on the phenotype could not be separately analyzed in this study.
The researchers also identified five amino acid substitution sites that may define new lineages and found possible evidence of positive selection pressures. Significantly, they found this to be true at the site of the clade B defining mutation NS1-M349V, this clade showing high replicative fitness in human cells.
The separation of the fitness experiments in human cells and mosquitoes ruled out the possibility of analysis of the changes in replicative fitness occurring over a natural cycle of transmission. However, they did allow the study of the complete course over which new lineages emerged in these two organisms. Some of these lineages may thus be relevant in natural transmission as well, especially since the two which have increased replicative fitness have no negative effects in either line.
On the other hand, many phenotypic alterations specific to certain lineages were observed to arise throughout the Zika epidemic, but few were fixed. The two lineages with increased fitness in one host may have lower fitness in the other, limiting their survival and transmission. Moreover, the lack of envelope mutations in all 13 major lineages identified in this study suggests that immune evasion or escape variants have not taken root during this epidemic.
What are the implications?
Two lineages appeared during mosquito and human cell infection. The mechanism for the emergence of the five lineages during this period seems to be adaptive evolution. Interestingly, this did not lead to the rise of fitter lineages displacing the less fit ones. Future studies should look into the results of the mutations defining these lineages regarding their phenotypic effects.
The fundamental importance of these lineages in Zika virus survival and transmission in nature remains to be studied because of the lab-based nature of the experiments in this study. However, the strong agreement between the differences in replicative fitness among all the human cell lines infected with the virus suggests that these effects may hold well over a broad spectrum of human tissues.
“Taken together, our findings suggest that while Zika virus likely acquired phenotypic changes during the 2015–2016 epidemic as it evolved in response to novel environments in the Americas, it is unlikely these changes had a significant impact on the course of the epidemic.”
The study provides a screening framework for differences in fitness over an epidemic and suggests several changes in viral phenotype during this period, though without significant effect on the course of the outbreak. If such changes can be kept under surveillance at such times, the disease spread and severity could be mitigated more easily.
“We believe this framework can be applied to study phenotypic evolution during future epidemics caused by emerging RNA viruses.”


